Novae: a graph-based foundation model for spatial transcriptomics data
Preprint on bioRxiv
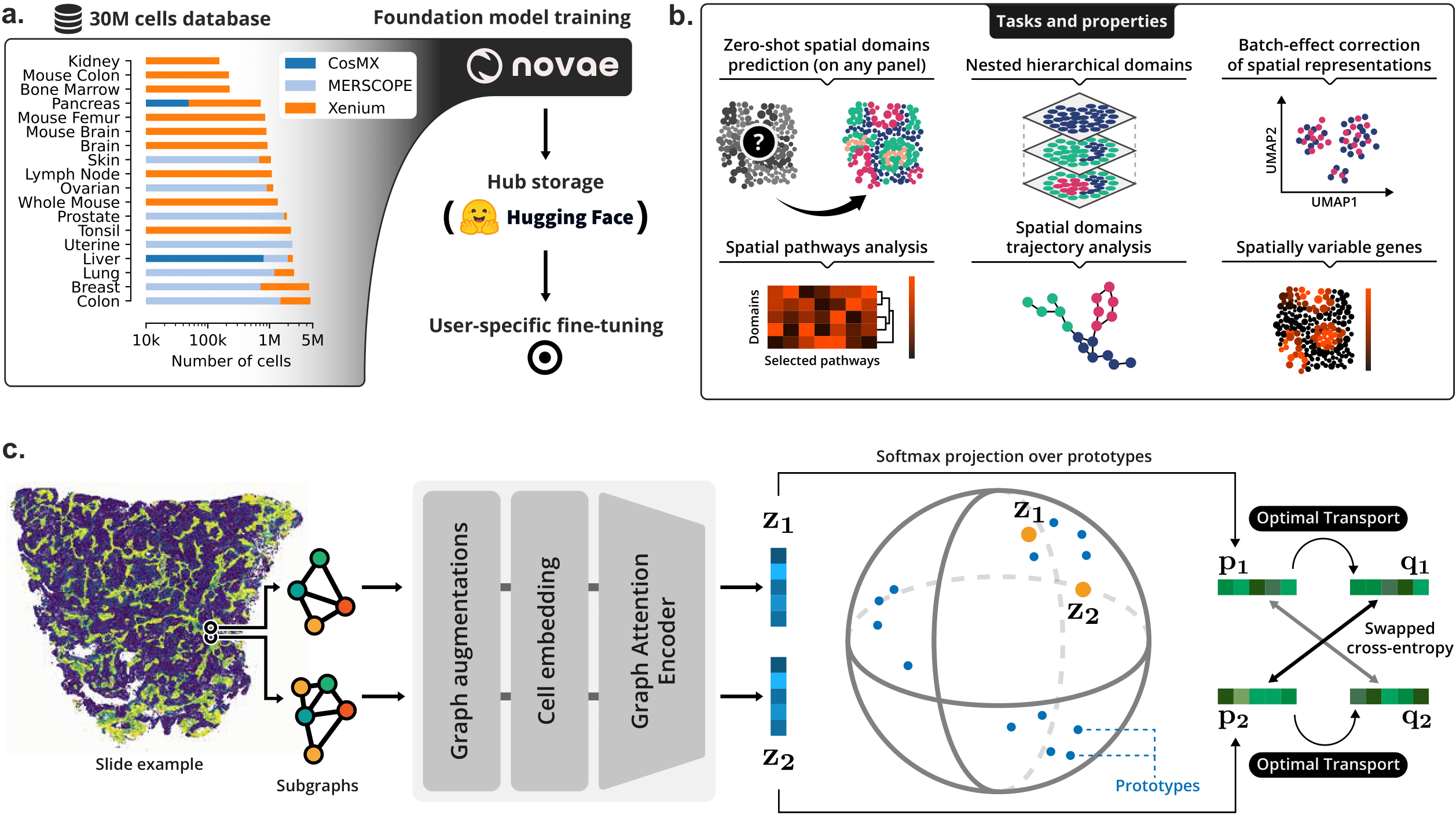
Spatial transcriptomics is advancing molecular biology by providing high-resolution insights into gene expression within the spatial context of tissues. This context is essential for identifying spatial domains, enabling the understanding of micro-environment organizations and their implications for tissue function and disease progression. To improve current model limitations on multiple slides, we have designed Novae (https://github.com/MICS-Lab/novae), a graph-based foundation model that extracts representations of cells within their spatial contexts. Our model was trained on a large dataset of nearly 30 million cells across 18 tissues, allowing Novae to perform zero-shot domain inference across multiple gene panels, tissues, and technologies. Unlike other models, it also natively corrects batch effects and constructs a nested hierarchy of spatial domains. Furthermore, Novae supports various downstream tasks, including spatially variable gene or pathway analysis and spatial domain trajectory analysis. Overall, Novae provides a robust and versatile tool for advancing spatial transcriptomics and its applications in biomedical research.